Although papaverine is not under international narcotics control it is an opium alkaloid and so within the interest of readers of the Bulletin. Medical use has created a demand in excess of the supply obtained from opium. This article reviews the methods of synthesizing this alkaloid
Author: Dov Elad, David Ginsburg
Pages: 27 to 34
Creation Date: 1952/01/01
Although papaverine is not under international narcotics control it is an opium alkaloid and so within the interest of readers of the Bulletin. Medical use has created a demand in excess of the supply obtained from opium. This article reviews the methods of synthesizing this alkaloid
Papaverine (1-(3,4-dimethoxybenzyl)-6,7-dimethoxyisoquinoline) is a member of the benzylisoquinoline sub-group of the opium alkaloids and is present in most varieties of opium to the extent of 0.5 to 1 per cent. Since the alkaloid is one of nature's useful antispasmodics, it soon found its way into the physician's arsenal, and serves in cases when it is necessary to cause relaxation of smooth muscle. Papaverine, in the form of its hydrochloride, is administered intravenously in treating pulmonary arterial embolism and orally or by injection in treating renal or biliary colic. It is not effective in abolishing neurally excited spasms and has undesirable side-effects, such as causing prolonged fall in arterial blood pressure, when the intestinal tract is relaxed. This is due to lack of selectivity in relaxing smooth muscle. Despite such undesirable side-effects, nature does not produce enough papaverine to meet the demands of medical practice, and many syntheses have been devised for preparation of this alkaloid. It is the purpose of this article to review the published methods for the production of synthetic papaverine.
A short account of the researches leading up to the assignment of the structure of papaverine, would, perhaps, be in order. Papaverine was first isolated by G. Merck,[1] in 1848, who assigned to it the correct empirical formula C 20H 21O 4N. The structure of the alkaloid was determined mainly by Goldschmiedt and his co-workers.[2] Some confusion existed in the early work through the misconception that a quinoline nucleus was present in papaverine. It was later shown,[3] that an isoquinoline nucleus is present.* The most important data regarding the structure were obtained by means of oxidative degradation.
* See VII.
Papaverine, upon oxidation with permanganate under different conditions of pH, yields a variety of degradation products. Among these are 6,7-dimethoxyisoquinoline-1-carboxylic acid (I), 6,7-dimethoxyisoquinoline (II), metahemipinic acid (III), and cinchomeronic acid (IV).
The formation of these products is consistent with the following formulation:
The point of attachment of the veratryl residue was demonstrated by oxidation with hot permanganate. Under these conditions, pyridine-2, 3, 4-tricarboxylic acid (V) was isolated. The veratryl residue appeared in the oxidation mixture in the form of veratric acid (VI). Structure VII was therefore assigned to papaverine.
Since this structure is relatively simple, it was soon proved by synthesis. A number of improvements have been made since the original synthesis, which considerably simplify the preparation of papaverine. Synthetic papaverine has out-distanced the naturally occurring alkaloid in volume of production.**
**The reasoning applied in correcting the early misconception is of great interest but is beyond the scope of this article. For a discussion, see reference 4. References will be found at the end of this article.


The first synthesis of papaverine was accomplished by Pictet and Gams.[5] The synthetic sequence is shown in chart I.
The two intermediates in this synthesis are 3,4-dimethoxy-ω-amino-acetophenone (XI; chart I, (a)) and homoveratroyl chloride (XVI; chart I, (b)).
3,4-dimethoxy-acetophenone (IX) was obtained through a Friedel-Crafts reaction of veratrole (VIII) with acetyl chloride. This ketone was converted through the oximino-ketone X to the corresponding ω-aminoacetophenone XI by reduction with stannous chloride.
Veratric aldehyde (XIII) was obtained by methylation of vanillin (XII). The cyanohydrin XIV was prepared and upon treatment with hydriodic acid, several transformations took place: demethylation of the phenolic ether groups, saponification of the nitrile group and reduction of the secondary hydroxyl group. Remethylation of the product gave homoveratric acid (XV) which was converted to its acid chloride (XVI) by means of phosphorus pentachloride.
Condensation of the hydrochloride of 3,4-dimethoxy-ω-amino-acetophenone (XI) with homoveratroyl chloride (XVI) yielded the β-keto-amide XVII. Reduction with sodium amalgam gave the corresponding β-hydroxy-amide XVIII. Dehydration of XVIII with phosphorus pentoxide at the reflux temperature of xylene yielded papaverine (VII).
Pictet and Finkelstein[6] improved the synthesis by substituting β-(3,4-dimethoxyphenyl)-ethyl amine (XIX) for the above ω-amino-acetophenone. Condensation of this amine with homoveratroyl chloride yielded the amide XX. Cyclization of the amide by the Bischler-Napieralski procedure (phosphorus oxychloride), yielded 3,4-dihydropapaverine (XXI). It remained to Spath and Burger[7] to accomplish the dehydrogenation of this substance, by treatment with palladized-asbestos at 200° in the presence of air. The latter workers showed also that 1, 2, 3, 4-tetrahydropapaverine (XXXIII) could be dehydrogenated to papaverine by means of the same catalyst. The reactions discussed are summarized in chart II.

Two groups, headed by Rosenmund and Mannich, respectively, employed 3,4-dimethoxy-ω-nitro-styrene (XXII) as an intermediate in the synthesis of papaverine.8, 9 This approach makes use of the observation that alcohols can be added to the double bond of ω-nitro-styrenes and is summarized in chart III.
The ω-nitro-styrene XXII is obtained by alkaline condensation of veratric aldehyde (XIII) with nitromethane. The methyl ether XXIII is obtained by the addition of the elements of methanol to the double bond of XXII. Reduction either with zinc-copper alloy in the presence of formic acid or with sodium amalgam in the presence of acetic acid, gives the methyl ether of the corresponding amine (XXIV). Treatment of the latter with homoveratroyl chloride yields the methyl ether XXV of the β-hydroxy-amide (XVIII) which is an intermediate in the original Pictet-Gams synthesis. In analogy to that synthesis (in which two moles of water were removed in the final cyclization step), one mole water and one mole methanol are removed by a cyclization agent, and papaverine is formed.



Buck[10] dehydrated the ,β-keto-amide which had previously been obtained as an intermediate in the Pictet-Gams synthesis, by means of phosphorus oxychloride, and formulated the product as XXVI. This was reduced with a palladium-platinum catalyst to a product formulated as XXVII and the latter was dehydrated by means of phosphorus pentachloride in chloroform to a crystalline substance formulated as 1,2-dihydropapaverine (XXVIII). That this crystalline compound was, indeed, a dihydropapaverine was proved by dehydrogenation to papaverine.
Young and Robinson,[11] in a brilliant analysis of the properties of Buck's intermediates, showed that these were incompatible with the above formulation (XXVI to XXVIII). They showed that the product formulated as XXVI was, in fact, an oxazole, XXIX, which is reducible to the amide obtained previously by Pictet and Finkelstein[6] (XX); furthermore, the dihydropapaverine obtained was a purer sample of 3,4-dihydropapaverine than had been previously isolated as an oil. Mixed melting point determinations showed that Robinson's reasoning was correct and that Buck had synthesized, 3,4-dihydropapaverine. The 1,2-dihydroisomer has not been prepared.
Spath and Berger[12] synthesized papaverine in low yield by a route which is of interest because it employs intermediates which are likely to be the biogenetic precursors of the alkaloid. Their method is shown in chart IV.
Ozonization of eugenol methyl ether (XXX), yielded 3,4-dimethoxyphenyl-acetaldehyde (XXXI). When this biogenetically-feasible intermediate was condensed with β-(3,4-dimethoxyphenyl)-ethyl amine (another substance capable of being available in the plant) the Schiff base XXXII is obtained. Upon heating with hydrochloric acid, this gave a low yield of 1, 2, 3, 4-tetrahydropapaverine (XXXIII), which, as mentioned previously, can be dehydrogenated to papaverine. Though this synthesis has no practical value, it may indicate the biogenetic route (albeit under much milder conditions) of the synthesis of papaverine in the plant.
Kindler and Peschke[13] introduced several important practical modifications in the Pictet-Gams synthesis and improved the over-all yield of papaverine. They subjected 3,4-dimethoxy-acetophenone (IX) to the conditions of the Willgerodt reaction (treatment with sulphur and dimethyl amine) and obtained homoveratryl N-dimethyl thio-amide (XXXIV). The latter substance was hydrolyzed with potassium hydroxide to homoveratric acid (XV).
Moreover, these workers found that the acid could be condensed directly with β-(3,4-dimethoxyphenyl)-ethyl amine (XIX) by heating the components in tetralin solution with simultaneous distillation of the water formed in the reaction. This method gives a nearly quantitative yield of the Pictet-Finkelstein[6] intermediate XX. The latter was submitted to cyclization with phosphorus oxychloride and dehydrogenation of the resulting 3,4-dihydropapaverine was accomplished by palladium in boiling dihydrophellandrene which is claimed to be an excellent medium for this reaction.

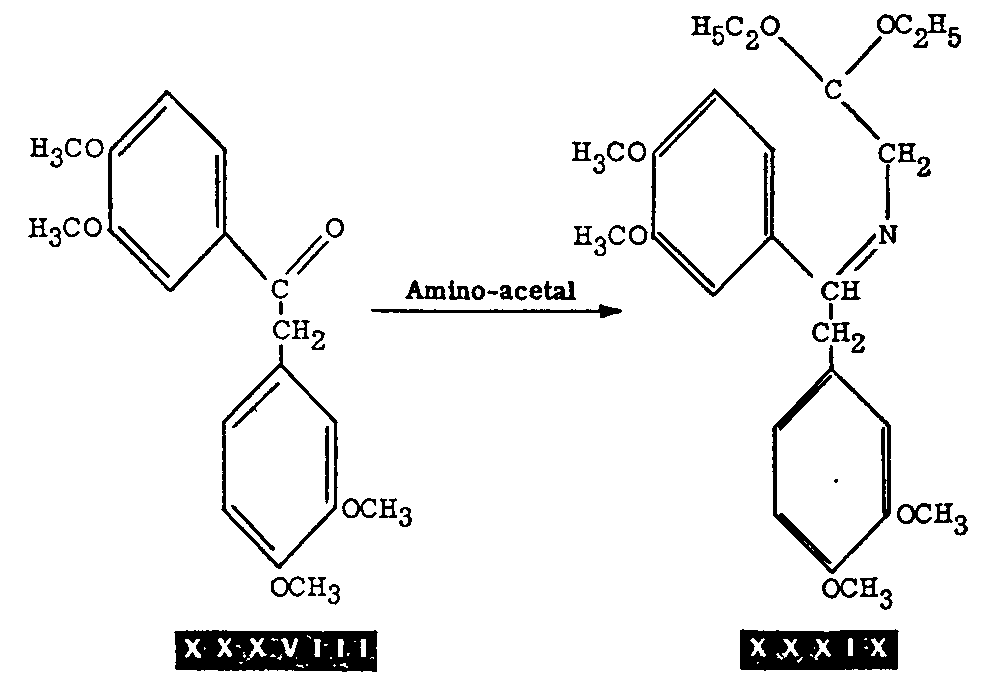
Papaveraldine (1-(3,4-dimethoxybenzoyl)-6,7 - dimethoxy-isoquinoline) (XXXVI) has been synthesized by Buck, Perkin, and Stevens.[14] Its synthesis is formally a synthesis of papaverine since it has been shown[15] that papaveraldine can be reduced to papaverine through the intermediate carbinol-papaverinol (XXXVII).*
In this synthesis, 3,4-dihydropapaveraldine (XXXV) is obtained by air oxidation of 3,4-dihydropapaverine (XXI) and papaveraldine (XXXVI) is obtained by treatment of the dihydro-compound with alcoholic potassium hydroxide in the presence of air. These transformations and the conversion of papaveraldine to papaverine are summarized in chart V.
*This synthesis is recorded here because it is formally a synthesis of papaverine. Obviously, from the practical point of view, 3,4-dihydropapaverine is directly dehydrogenated to papaverine. This synthesis illustrates, however, the great susceptibility of 3,4-dihydropapaverine to air oxidation. For this reason, cyclization of XX to yield XXI and dehydrogenation of XXI are carried out in an inert atmosphere, in plant practice.
A number of unsuccessful attempts to synthesize papaverine has been recorded in the literature.

Fritsch[16] condensed amino-acetal with desoxy-veratroin (XXXVIII) and cyclized the product (XXXIX) with concentrated sulphuric acid. Instead of obtaining papaverine, a substance melting 15° higher was formed, the structure of which was not elucidated. Similarly, Allen and Buck,[17] in an attempt to extend Rugheimer and Schon's synthesis of 6,7-dimethoxyisoquinoline,[18] reduced the oxime of desoxyveratroin (XL) with sodium amalgam, condensed the resulting amine XLI with bromoacetal and obtained XLII. Upon attempted cyclization of the latter substance, however, decomposition occurred and no papaverine isolated.


Kefford[19] intended to prepare papaverine by condensation of XLIII with the Grignard reagent from homoveratryl bromide (XLIV). The intermediate  expected, XLV, would then undergo intramolecular cyclization as indicated in the formula. This approach failed, however, when the desired Grignard reagent could not be prepared.
expected, XLV, would then undergo intramolecular cyclization as indicated in the formula. This approach failed, however, when the desired Grignard reagent could not be prepared.

Syntheses of various substances which can be used as intermediates in the synthesis of papaverine have been described. Many isoquinolines resembling papaverine in structure have been synthesized and, undoubtedly, reactions which have been utilized in the preparation of their intermediates can be applied to the preparation of papaverine intermediates. However, full details of processes for the production of synthetic papaverine currently employed by pharmaceutical manufacturers have not been disclosed.
Certain obvious routes for the synthesis of important papaverine intermediates may now be summarized.
A common precursor for both β-(3,4-dimethoxyphenyl)-ethyl amine and homoveratric acid, is 3,4-dimethoxyphenylacetonitrile (XLVIII). This nitrile can be prepared, for example, as shown in chart VI.
Veratraldehyde (XIII) can be converted to veratryl alcohol (XLVI) by catalytic reduction,[20] by reduction with aluminium isopropylate,[21] or by means of a cross-Canizzaro reaction with formaldehyde[22] The chloride XLVIIa can be obtained by treatment of the alcohol with hydrogen chloride or thionyl chloride. Similarly, the bromide XLVIIb can be obtained from the alcohol by treatment with hydrogen bromide[23] Either halide is converted to the nitrile XLVIII by means of potassium cyanide.21, 24
Reduction of the nitrile at low or high pressure in the presence of Raney nickel or at low pressure in the presence of noble metal catalysts, yields the required amine XIX. The yield of primary amine has been improved by carrying out the reduction in the presence of ammonia. On the other hand, saponification of XLVIII yields homoveratric acid (XV)[25]
An alternative preparation of 3,4-dimethoxybenzyl chloride (XLVIIa) by chloromethylation of veratrole (VIII) has been described.24, 26 This method is of practical importance in shortening the number of steps in the papaverine synthesis, and because cheap reagents are utilized in the chloromethylation reaction.
The preparation of β-(3,4-dimethoxyphenyl)-ethyl amine (XIX) in 80 per cent yield by palladium-black reduction of 3,4-dimetroxy-ω-amino-acetophenone (XI), has been reported.[27] The same catalyst effects the reduction of 3,4-dimethoxybenzoyl cyanide (XLIX) to the same amine in 72 per cent yield.[27] The reduction of ω-nitro-styrenes similar to XXII by means of aluminium amalgam,[28] lithium aluminium hydride,[29] or catalytically,[30] to substances analogous to XIX, has been reported. A synthesis of XIX from eugenol methyl ether (XXX) has been devised.[31] Oxidation of the latter with neutral permanganate solution yields the glycol L. The glycol was oxidized with lead tetraacetate to give 3,4-dimethoxyphenylacetaldehyde (XXXI). The oxime of this aldehyde was then reduced to XIX.


It may be seen from the above discussion, that the synthesis of the key substances in the synthesis of papa- verine (XV and XIX) has been rather exhaustively investigated. The methods actually used in production of synthetic papa verine today, combine steps giving the best yields and utilizing the most favourable reaction conditions. Through the improvements listed above, the synthesis of papaverine has been considerably shortened and the yield of the alkaloid substantially increased in the four decades following the first synthesis of this important antispasmodic by Picket and Gams.
Merck, Ann., 66, 125, (1848); ibid., 73 , 50 (1850).
002Goldschmiedt and co-workers, Monatsch, 4, 704, (1883); ibid, 6 , 372, 667, 954 (1885); ibid., 7 , 485 (1886); ibid, 8 , 510 (1887); ibid., 9 , 42, 327, 349, 679, 762, 778 (1888); ibid, 10 , 156, 673, 692 (1889); ibid, 13, 697 (1892); ibid., 17 , 491 (1896); ibid., 19 , 321 (1898); ibid., 24 , 681 (1903); Ber. 36 , 1850 (1903).
003Claus, Ber., 16, 1284 (1883); Hoogewerff and Van Dorp, Rec. tray. chim, 4 , 285 (1885); Goldschmiedt, Monatsch., 9 , 327, 349 (1888).
004Small, Chemistry of the Opium Alkaloids, pp. 3-6, U.S. Govt. Printing Office, Washington, 1932.
005Pictet and Gams, Ber, 42, 2943 (1909); C. R. Ac Sc, 149, 210 (1909).
006Pictet and Finkelstein, Ber, 42, 1979 (1909); C. R. Ac Sc. , 148 , 925 (1909).
007Spath and Burger, Ber., 60, 704 (1927).
008Rosenmund, Nothnagel, and Riesenfeldt, ibid, 60, 392 (1927).
009Mannich and Walther, Arch. Pharm., 265, 1 (1927).
010Buck, J Am. Chem. Soc., 52, 3610 (1930).
011Young and Robinson, J. Chem. Soc., 1933 , 275.
012Spath and Berger, Ber., 63, 2098 (1930).
013Kindler and Peschke, Arch. Pharm, 272, 60, 236 (1934).
014Buck, Haworth, and Perkin, jr., J. Chem. Soc., 125, 2176 (1924).
015Buck, Perkin, jr., and Stevens, ibid., 127, 1462 (1925).
016Fritsch, Ann., 329, 37 (1903).
017Allen and Buck, J. Am. Chem. Soc, 52 , 310 (1930).
018Rugheimer and Schon, Ber., 42 , 2374 (1909).
019Keflord, J. Chem. Soc., 1940 , 1209.
020Kindler and Gehlhaar, Arch. Pharm., 274 , 377 (1936).
021C f . Schopf and Salzer, Ann., 544 , 14 (1940); Schopf and Winterhalder, ibid, 544 , 71 (1940).
022Davidson and Bogert, J. Am. Chem. Soc., 57, 905 (1935).
023Woodward, J. Am. Chem. Soc., 62 , 1481 (1940).
024Bide and Wilkinson, J. Soc. Chem. Ind., 64 , 84 (1945).
025Wenner, J Org. Chem., 15 , 548 (1950).
026Gawron, J. Am. Chem. Soc., 71 , 744 (1949).
027Kindler, Hedemann and Scharfe, Ann., 560 , 215 (1948).
028C f . Spath, Riedl, and Kubiczek, Monatsch., 79, 72 (1948).
029C f . Hamlin and Weston, J. Am. Chem. Soc, 71 , 2210 (1949); Ramirez and Burger, ibid., 72 , 2781 (1950); Gins-burg, Bull. soc. chim. France (5), 17, 510 (1950).
030Cf. Kindler, Brandt, and Gehlhaar, Ann., 511, 209 (1934)
031Kaufman, Eliel, and Rosenkranz, Ciencia, 7, 136 (1946); Chem. Abstr, 41, 2398 (1947).
Addendum
A new synthesis of papaverine has been reported independently by Wahl[32] and by Galat.[33] In this approach a 3-carbosy-3,4-dihydropapaverine (LVI), is the intermediate which Wahl decarboxylated and dehydrogenated in one step, while Galat isolated the intermediate 3,4-dihydropapaverine (XXI) and then dehydrogenated this in the usual way. The yield of the carboxy-amide LIII is higher when LII is treated with ammonia than when LI is treated with the same reagent. Esterification of the free acid LIII followed by phosphorus oxychloride cyclization of the methyl ester LIV yields the 3-carbomethoxy-3,4-dihydropapaverine (LV). Dehydrogenation of this compound is known to give 3-carbomethoxypapaverine.[34] The ester LV was therefore saponified and the free acid obtained (LVI) converted to papaverine either directly or stepwise, as indicated above.
The reactions involved are summarized in chart VII on page 34.
For the sake of completeness, the reviewers are including in this section an interesting report by Eistert,[35] on the synthesis of the key intermediate XX, despite the lack of experimental details in the report. The diazoketone LVII, upon treatment with silver oxide in the presence of ?-(3,4-dimethoxyphenyl)-ethyl amine (XIX), evidently forms the amide XX directly. See chart VIII on page 34.
References
032Wahl, Bull. soc chim. France (5), 17, 680 (1950).
033Galat, J Am Chem. Soc., 73, 3654 (1951).
034Redel and Bouteville, Bull. soc. chim. France (5), 16, 443 (1949).
035Eistert, Angew Chem, 54, 124 (1941).



